Small and medium size medical device companies
By May 2020 medical devices companies will have to comply with the new EU Medical Device Regulation (MDR). For Vitro Medical Devices (IVMD) the date is May 2022.
The regulation holds several new and/or increased requirements that must be adhered to:
- Implementation of a Quality Management System, with a Conformity Assessment
- New classification system supported by the Conformity Assessment
- Requirements for CE marking on the device supported by technical documentation
- Compliance with reporting of clinical investigations and post-market surveillance requirements
- Appointing an Authorized Representative and/or a Regulatory Responsible
In cooperation with MyBlueLabel, Pharma IT can now offer a turn-key solution and services that will enable small and medium size medical device companies to comply with new regulations listed above.
Larger or more complex medical devices companies, pharmaceutical companies and biotech companies might benefit from other solutions, as such Pharma IT work as an agnostic system advisor.
The Pharma IT turn-key solution and services can be delivered individually or as a package deal. In this article we have listed the minimum required effort to implement our solution assuming companies will do more work on their own, but additional services can be provided by Pharma IT on an ad-hoc basis, see figure below.

Quality Management System (QMS)
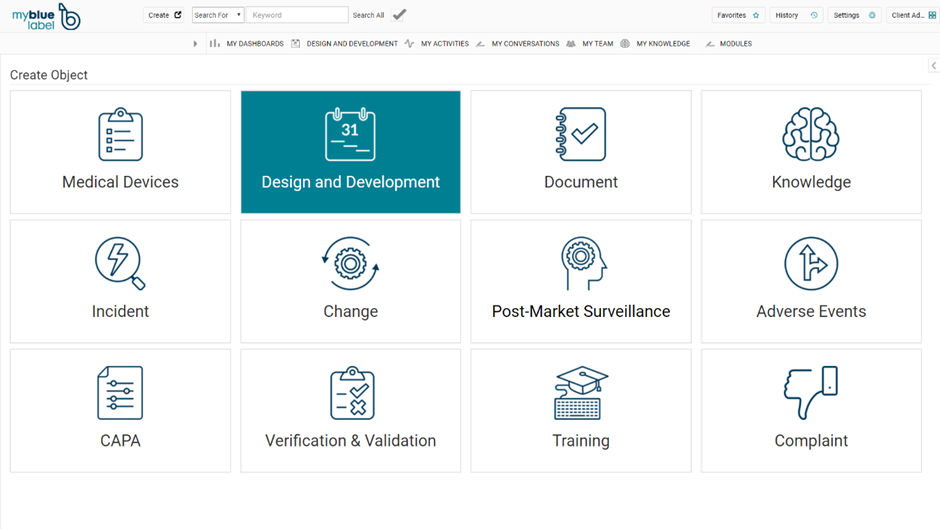
In corporation with MyBlueLabel, Pharma IT can offer a simple electronic document management system and quality management system that contains all the functionality needed to comply with the requirements of the new MDR and IVDR regulation. The solution only costs 470 EUR/medical device/month and as outlined in the figure below the system contains functionality to support the following:
- Management of Medical Devices
- Management of Design and Development of Medical Devices
- Document Management and electronic signatures for review and approval of documents
- Management of Obtained knowledge and lessons learned
- Management of Quality Incidents
- Management of changes
- Management of Medical Device Adverse Events
- Management of Corrective and Preventive Actions
- Management of Test and Validation of IT systems or medical devices
- Management of Training of Employees
- Management of Customer Complaints and Complaints in general
Furthermore, we can offer a set of document templates that are compliant with ISO9001 and ISO13485 which can be used as a basis for creating company specific policies and procedures to comply with the new regulation. The template package includes a Quality Manual template, templates for standard operating procedures (see figure below) as well as IT system lifecycle management procedures to ensure compliance of the electronic document management system and quality management system over time.
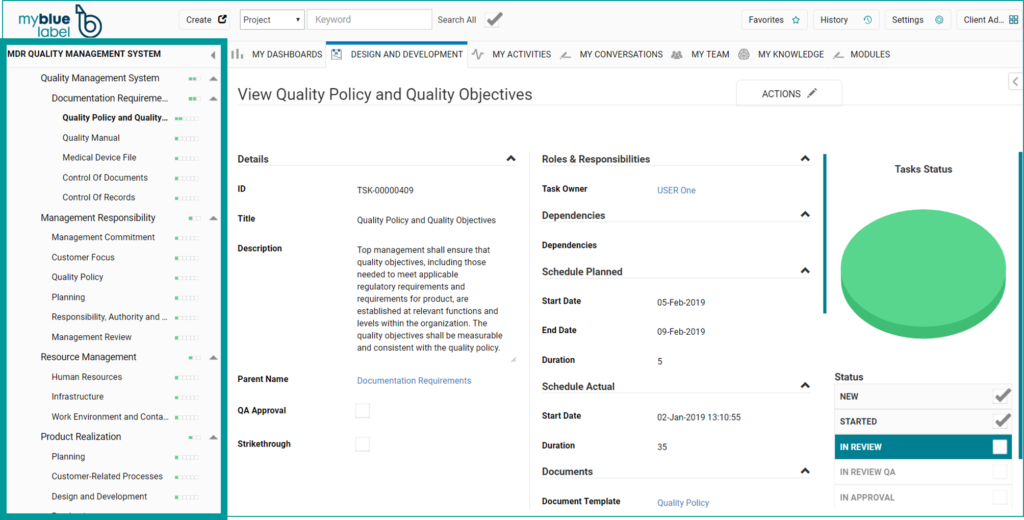
If all document templates are purchased the package costs 13.500 EUR, if you as a company have some procedures in place and only want to purchase a limited set of templates the price can be reduced.
For the full implementation of the MyBlueLabel solution Pharma IT recommends including support for adjustment and extended training of the QMS to specific company standards (3 days), validation of the IT system (3 days) and end user training of the company in the use of the system (2 days). These services are optional and can also be performed by the company itself.
If more support is needed this can be provided on an ad-hoc basis and Pharma IT can also provide support for maintenance of the IT system after implementation.
Conformity Assessment
The document templates also come with a conformity assessment template that will guide you through the assessment of the device and thereby correct classification in respect to the new regulation.
Technical documentation
As part of the new regulation the medical device will have to be UDI and CE marked. The basis for the UDI and CE marking will be the technical documentation. The MyBlueLabel solution comes with an electronic document management system that can handle and manage the technical documentation for each Medical Device to ensure that you will obtain the benefits of electronic document management:
- Reduced Storage Space
- Enhanced Security
- Improved Regulatory Compliance
- Easier Retrieval
- Better Collaboration
- Better Backup and Disaster Recovery
Compliance with reporting of clinical investigations and post-market surveillance requirements
The new regulation requires additional information in respect to reporting of clinical investigations, post-market surveillance in respect to post-market surveillance plan, PSURs, reporting of incidents and trending reports. Pharma IT have an experienced safety and pharmacovigilance team from the Pharmaceutical Industry who is experienced in this line of work and based on an individual company assessment we can deliver the right service for your specific needs.
Authorized Representative and Regulatory
Pharma IT can offer qualified personal which can be appointed both as Authorized Representative and Regulatory Responsible.
The authorized representative will be responsible for products on the market and will support your company with internal audits and reviews to ensure proper management of Quality. The Authorized Representative will be legally liable for defective devices on the same basis as, and jointly and severally with, as the manufacturer.
The Regulatory responsible will ensure compliance in respect to regulations from authorities. The person will be responsible for check of conformity of the devices in respect to the QMS before a device is released, check of the technical documentation and the EU declaration of conformity are kept up-to-date, check that the post-market surveillance obligations are performed and that the reporting obligations are fulfilled.
Please contact us for more information
Should you find our turn-key solution for becoming compliant with MDR and IVDR by May 2020/2022 interesting please contact us via this link.
If interested you can sign-up for free demo of the MyBlueLabel system and a 30 days free trial license.