Share on email
Share on linkedin
Share on twitter
Share on skype
Share on facebook
By Lena Brahe
November 21, 2022
By Lena Brahe
November 21, 2022
If you currently conduct or plan to conduct clinical trials in Europe, you will be impacted by the new EU Clinical Trials Regulation (CTR) that entered into application on 31 January 2022[1].
From January 31st, 2023, it will be mandatory to use CTIS for submission of clinical trial applications (CTAs) and for documents related to the maintenance of the clinical trial authorisation in Europe[2].
The EU CTR aims to provide more transparency on clinical trial information. This means that a number of documents submitted to CTIS will become public at the first opportunity, i.e., time of decision[3]. Sponsors can request to defer the publication of documents submitted to CTIS. However, this deferral must be requested at the time of the initial CTA.
Sponsors will need to redact personal and commercially confidential information before submitting documents in CTIS. The redaction may impact most of the documents submitted and will be a comprehensive task. Sponsors are therefore encouraged to create a redaction strategy and ensure resource allocation in due time.
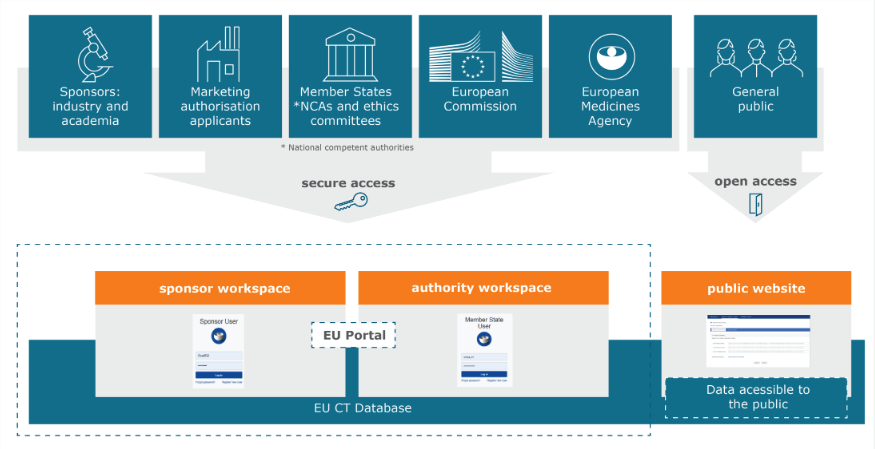
The Investigator’s Brochure (IB) is a “compilation of the clinical and nonclinical data on the investigational product(s) that are relevant to the study of the product(s) in human subjects” [5]. As per the new requirements put forth by the EU CTR, the IB must be submitted to CTIS and disclosed.
Given this, sponsors should evaluate their current process for development of IBs and take several considerations into account, including the following:
Increased document disclosure requirements is just one of the key changes that will come into affect with the new EU CTR. For an overview of the top 5 changes that you should be mindful of as a sponsor when conducting clinical trials in Europe you can read our previous Insight on the topic, here.
At Pharma IT, we have experience in assisting our customers with ensuring their current and future clinical trial strategy has incorporated the changes necessitated by the new EU CTR. We suggest all organizations taking a moment to think through the following:
If you could benefit from further assistance in planning and ensuring EU CTR compliance, do not hesitate to reach out to the Pharma IT team.
Sources Cited
[1] Clinical Trials Regulation | European Medicines Agency (europa.eu)
[2] Clinical Trials Information System | European Medicines Agency (europa.eu)
[4] CTIS Highlights Issue 1, June 2020 | European Medicines Agency (ema.europa.eu)
[5] Investigator’s Brochure: ICH E6 (R2) Good clinical practice (https://ichgcp.net/da/7-investigators-brochure)
Lena Brahe is a Principal Consultant at Pharma IT and holds more than 10 years’ experience in the pharmaceutical industry and academia working with Medical Writing, Clinical Science and Project Management. Lena has experience with clinical development programmes and regulatory processes, and has driven clinical documents including Clinical Study Protocols, Participant Information/Informed Consent forms, Investigator’s Brochures, Clinical Study Reports, Health Authority Meeting Packages, and CTD Submission Documents, in addition to Scientific Papers.

