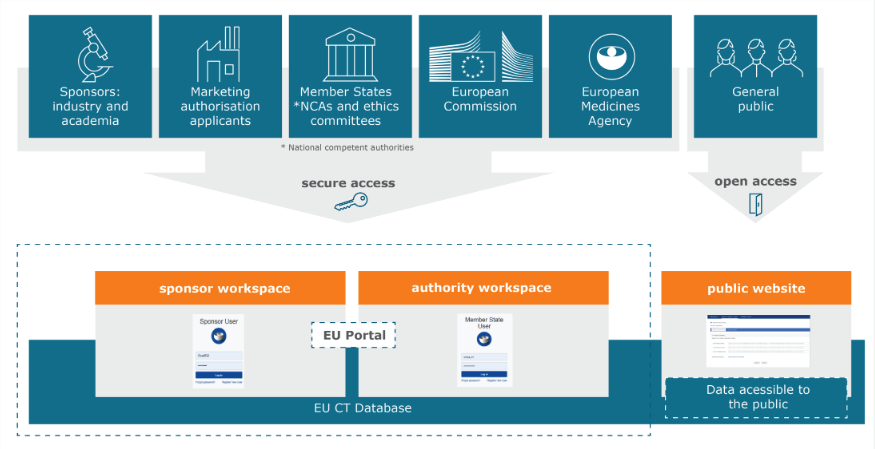
Increased document disclosure requirements is just one of the key changes that will come into affect with the new EU CTR. For an overview of the top 5 changes that you should be mindful of as a sponsor when conducting clinical trials in Europe you can read our previous Insight on the topic, here.
At Pharma IT, we have experience in assisting our customers with ensuring their current and future clinical trial strategy has incorporated the changes necessitated by the new EU CTR. We suggest all organizations taking a moment to think through the following:
- Which clinical trials are in scope and what will the impact be?
- What is mandatory for sponsors and what can be outsourced?
- How will the EU CTR impact your SOPs and processes?
- Which IT systems may be impacted?
If you could benefit from further assistance in planning and ensuring EU CTR compliance, do not hesitate to reach out to the Pharma IT team.
Sources Cited
[1] Clinical Trials Regulation | European Medicines Agency (europa.eu)
[2] Clinical Trials Information System | European Medicines Agency (europa.eu)
[3] Appendix on transparency rules to the Functional Specifications of the EU clinical trials portal and database (europa.eu)
[4] CTIS Highlights Issue 1, June 2020 | European Medicines Agency (ema.europa.eu)
[5] Investigator’s Brochure: ICH E6 (R2) Good clinical practice (https://ichgcp.net/da/7-investigators-brochure)